



管丽医生的科普号
- 精选 秋季腹泻来临,家长如何应对?
什么是秋季腹泻?秋季腹泻医学上特指由轮状病毒感染所致,由于轮状病毒喜冷怕热,多发于秋冬季,俗称为“秋季腹泻”。秋季腹泻多发生于3岁以内的儿童,以6个月到1岁的孩子多见。患儿往往起病急,开始表现为发烧、咳嗽、流清鼻涕等“感冒”症状,同时伴有频繁呕吐,呕吐物为胃内容物,腹泻少则一天数次,多则数十次,大便稀薄,呈清水样或蛋花汤样,有时呈白色米汤样,多无特殊腥臭味。实验室检查大便常规正常,但轮状病毒检测为阳性。如何治疗秋季腹泻?秋季腹泻虽为自限性疾病,但只有少部分症状较轻的患儿可不予治疗,因为腹泻会导致体内电解质丢失造成脱水,严重时甚至可危及生命。治疗上一般不采用抗菌素,除非患儿合并有细菌感染,主要是抗病毒和通过口服补液纠正脱水。腹泻次数多时可以口服蒙脱石散剂止泻,益生菌调节肠道菌群功能。秋季腹泻需要用抗生素吗?急性水样便腹泻病大部分为病毒感染,一般不用抗菌素。盲目使用抗菌素对孩子会导致体内正常菌群的紊乱。细菌感染或细菌病毒混合感染引起的腹泻病,如患儿血相高、大便检测有红或白细胞、脓细胞等,则需要应用抗菌素治疗。因此,应用抗菌素治疗腹泻病,要严格把握感染指针。怎么观察孩子有没有脱水?如果孩子腹泻次数频繁,呈水样、量多,尿量减少,皮肤弹性差,口唇干燥,甚至出现烦躁、嗜睡、萎靡、昏迷、惊厥等症状,大便出现粘冻血丝,前囟凹陷。孩子腹泻时如果有发烧、排脓血便、频繁呕吐、大便次数和量增多、精神不好等情况,一定要及时到医院诊治。孩子出现了脱水需要静脉补液吗?世界卫生组织推荐口服补液盐(Oral Rehydration Salts,ORS)治疗轻度脱水,这是将盐、葡萄糖、钾、水等尽量多喝水,水中可以加少量盐。腹泻严重时可以暂时禁食,但时间不宜太长。因为如果长时间只给孩子吃米汤等碳水化合物,饮食不均衡,会对孩子的生长发育造成影响,有可能导致营养不良,而且饥饿也会引起腹泻。如果护理患秋季腹泻的孩子?护理腹泻宝宝时家长要勤洗手,防止病从口入,防止病菌传播。避免孩子腹部受凉,因为腹部着凉时肠道蠕动加快,会导致腹泻。要注意孩子的腹部保暖,半夜醒来时看看孩子是否踢了被子。注意孩子小屁股的护理,由于腹泻时排出的粪便对皮肤刺激较大,频繁腹泻可能引起红臀,甚至肛周溃疡,因此要注意排便后臀部的清洗,小宝宝要勤换尿布,如果有肛周溃破要及时治疗。此外,要少带孩子去公共场所,避免交叉感染。孩子需要接种轮状病毒疫苗吗?6个月至5岁婴幼儿是秋季腹泻的易感人群,可选择接种轮状病毒疫苗。六个月到三岁以内孩子每年口服一次,三岁到五岁的孩子每年口服一次。在秋季腹泻流行季节接种轮状病毒疫苗预防效果更好。但不是每个孩子都可以接种疫苗,接种禁忌症包括:严重先天性疾病、过敏史、免疫缺陷者禁用和接受免疫抑制治疗者,孩子有发热、急性传染病或严重疾病时要暂缓接种,孩子在生病吃药时也不宜接种。本文系薛海虹医生授权好大夫在线(www.haodf.com)发布,未经授权请勿转载。
薛海虹 主任医师 上海新华医院 小儿内科7815人已读 - 精选 极长链酰基辅酶A脱氢酶缺乏症研究进展(发表于2011年国际儿科杂志)
极长链酰基辅酶A脱氢酶缺乏症研究进展 章瑞南(综述) 邱文娟(审校) 【摘要】 极长链酰基辅酶A脱氢酶缺乏症是一种较罕见的脂肪酸代谢障碍疾病,根据起病年龄和临床表现分为三型:心肌病型、肝型、肌病型。心肌病型病情重,死亡率高。临床诊断可通过血串联质谱(MS/MS)检测血肉豆蔻烯酰基肉碱(C14:1)水平进行,进一步确诊可通过基因诊断、酶学分析及脂肪酸氧化流量分析。治疗上主要包括避免空腹,减少长链脂肪酸的摄入,补充中链甘油三酯等。【关键词】 极长链酰基辅酶A脱氢酶(VLCAD) VLCAD缺乏症(VLCADD) 诊断 治疗Progresses in the study on Very long chain acyl-CoA dehydrogenase Deficiency ZHANG Ruinan,QIU wenjuan. Department of Pediatric Endocrinology,Genetic and Metabolic Diseases,Shanghai Institute of Pediatric Research,Xinhua Hospital,School of Medicine,Shanghai Jiaotong University,Shanghai 200092,China【Abstract】 Very-long-chain acyl-CoA dehydrogenase Deficiency(VLCADD) is a rare recessively inherited disorder of mitochondrial fatty acid β-oxidation. VLCADD is classfied into three types according the onset age and clinical manifestation: cardiomyopathic phenotype, hepatic phenotype and myopathic phenotype. The cardiomyopathic phenotype is most severe resulting in high mortality. The biochemical hallmark of the disease is elevation of C14:1-carnitine detected by tandem mass spectrometry (MS/MS). Enzyme analyses, molecular genetic analyses and fatty acid oxidation flux assay are used to make further diagnostic evaluation. Treatment regimens include avoidance of fasting, restriction of long-chain fat acid and supplementation of medium-chain triglycerides.【Key words】 Very-long-chain acyl-CoA dehydrogenase (VLCAD); VLCAD deficiency; diagnosis; treatment细胞内线粒体脂肪酸β氧化为机体多个器官和组织提供能量来源,脂肪酸氧化障碍是指脂肪酸β氧化过程中所需酶的功能障碍导致其氧化受阻、供能障碍及中间代谢产物蓄积的一组疾病。极长链酰基辅酶A脱氢酶缺乏症(Very long chain acyl-CoA dehydrogenase Deficiency, VLCADD,OMIM #201475)是由于细胞线粒体内脂肪酸β氧化中的关键酶极长链酰基辅酶A脱氢酶(Very long chain acyl-CoA dehydrogenase, VLCAD)基因先天缺陷所致的常染色体隐性遗传疾病,是一种较罕见的遗传代谢性疾病。近年串联质谱(tandem mass spectrometry, MS/MS)和气相质谱(gas-chromatography mass spectrometry,GC/MS)的运用对于该病的诊断起到很大的作用。本文将对该病的临床表现,分子病理机制,实验室检查和治疗等方面作一综述。1 概述极长链酰基辅酶A脱氢酶缺乏症最早由Bertrand于1993年报道[1]。VLCAD作为线粒体脂肪酸β氧化过程第一步的关键酶,催化含14个碳-18个碳的不同长度碳链的脂酰基辅酶A脱氢,其辅酶为黄素腺嘌呤二核苷酸(FAD),由FAD接受脱氢产生的氢原子进入线粒体呼吸链进行氧化磷酸化产生ATP供能。VLCAD在肝脏、心肌、骨骼肌、皮肤成纤维细胞的线粒体中均有表达,可催化长链酯酰辅酶A产生烯酯酰辅酶A,再在烯酯酰辅酶A水化酶、羟酯酰CoA脱氢酶、酮酯酰CoA硫解酶这三个酶的作用下共同完成长链脂肪酸的β氧化过程,每次生成一个乙酰辅酶A和少2个碳原子的脂酰辅酶A。乙酰辅酶A可参与三羧酸循环进行氧化磷酸化供能,也可在肝脏形成酮体,在运动、饥饿、应激等情况下产生能量。VLCAD缺陷将导致体内长链脂肪酸代谢障碍,长链脂肪酸不能氧化供能,同时蓄积在细胞内对心肌、骨骼肌、肝脏等产生毒性作用,导致VLCADD一系列的临床症状和体征。VLCADD发病率不确切,以肉豆蔻烯酰基肉碱(C14:1)作为新生儿VLCADD筛查指标的串联质谱新生儿疾病筛查显示各国发病率有明显差异:澳大利亚估计为1/31500[2],德国为1/125000,英国为1/42500[3]。Lindner等[4]统计澳大利亚、德国和美国约5250000新生儿筛查资料认为VLCADD发生率约为1/85000。上海市儿科医学研究所对278775新生儿筛查的串联质谱遗传代谢病筛查并未发现VLCADD患者[5];Shigematsu Y等[6]对日本102200新生儿筛查的串联质谱遗传代谢病筛查亦未发现VLCADD患者,提示亚洲国家本病罕见。但Tanmoki等[7]发现1985-2000年间日本187所大型医疗机构检出的64例脂肪酸氧化障碍中8例为VLCADD(12.5%),上海市儿科医学研究所对3070例遗传代谢病高危患者筛查所检出的13例脂肪酸氧化障碍中3例VLCADD(23.1%)[8],后续检出的共83例脂肪酸氧化障碍中11例为VLCADD(13.3%)。2 临床表现与分型VLCADD的临床表现有明显异质性,根据起病年龄和临床表现可分为3个类型[9-11],最常见的一种类型主要在新生儿和婴儿早期发病,常有心肌受累,又称心肌病型,此型发病凶险,患儿死亡率高,表现为低血糖、瑞氏综合征、新生儿猝死、肥厚型和扩张型心肌病、心包积液、心率失常、肌酸激酶水平升高;另外两种类型为轻型,包括婴儿后期或儿童发病的肝型和青少年或成年发病的肌病型。肝型患儿常表现为反复发作的低酮性低血糖,肝功能异常,很少伴有心肌损害,但未经及时诊断和治疗也会有生命危险。肌病型主要在青少年至成年期发病,为迟发型,症状轻,一般不伴有心肌疾病和低血糖,主要表现为运动、感染或饥饿后的横纹肌溶解和肌红蛋白尿,甚至可发生肾功能衰竭,可伴有肌无力,肌肉痛性痉挛或肌痛。Andresen等报道的55例VLCADD患者中,25例为心肌病型,占总数的46%,多在出生3天内发病(76%),伴有心肌病(92%),肝大(80%),肌无力(52%),预后较差,20例(80%)早期死亡,提示发病时间越早,病情越严重。55例中的21例为肝型,占总数39%,此型在儿童各期均有发病,但以4岁以内发病较多,伴有肝大(62%),肌无力(62%),低酮性低血糖(76%)。8人为肌病型,占总数的15%,均在13岁后发病,几乎所有患者有横纹肌溶解和肌红蛋白尿,伴有心肌病和肌无力的各占13%[9]。3 VLCADD的分子病理学机制极长链酰基辅酶A脱氢酶(VLCAD,EC 1.3.99.13) 由ACADVL基因(OMIM 609575)编码,ACADVL基因位于染色体17p13.1[12],长约5.4kb,含20个外显子,编码655个氨基酸前体蛋白,其中前40个氨基酸为前导肽,后615个氨基酸为成熟的多肽,分子量约为67KD,位于线粒体的内膜[13],属于脂酰辅酶A脱氢酶(ACADs)家族成员之一,VLCAD为同源二聚体的线粒体膜蛋白,而其他ACADs为线粒体基质的同源四聚体蛋白。VLCAD除了有与其他ACADs类似的N端400个氨基酸所形成的α螺旋-β折叠-α螺旋结构外,还另外具有C端180个氨基酸残基形成一个α螺旋将VLCAD蛋白锚定在线粒体内膜中,以类似于ACADs四聚体中二聚体间的作用方式与N端的两个α螺旋相互作用[14]。VLCAD与短链酰基辅酶A脱氢酶(SCAD)、中链酰基辅酶A脱氢酶(MCAD)、长链酰基辅酶A脱氢酶(LCAD)在线粒体脂肪酸β氧化过程中共同发挥作用,催化不同碳链长度的脂肪酸脱氢。VLCAD的N端400个氨基酸与SCAD、MCAD、LCAD等其他ACADs家族具有30%的同源性,且VLCAD的96位谷氨酸至480位赖氨酸包含脂酰辅酶A脱氢酶的结构域,此区域的突变可能影响酶的结构,可能导致突变蛋白的错误折叠以致降解[12]。462位谷氨酸为VLCAD的酶活性中心,其他ACADs该位点也高度保守,突变可导致酶活性完全丧失;C端与VLCAD在线粒体内膜基质侧结合功能有关,以481-516位点区影响最大,体外膜结合试验发现此区域突变可使VLCAD不能结合于线粒体内膜,但携带此区域的突变导致的表现型较轻,可能与此区域的突变仅影响酶的定位,而对酶活性的影响较小有关[14]。VLCAD的编码基因ACADVL的突变谱高度异质,迄今为止,已有116种突变类型报道,尚未发现某一突变为热点突变,其中错义突变为主要的突变类型,约占总突变的57%,缺失突变约占21%,剪切突变约占11%[9, 15-17]。多个研究认为极长链脂肪酸脱氢酶缺乏症患者的基因型与表现型之间存在明显相关性[9, 16],Andresen等研究发现71%的心肌病型患儿基因突变为无义突变;而在肝型和肌病型的基因突变中,分别有82%和93%的基因突变为错义突变或单个氨基酸缺失。无义突变可导致VLCAD完全失去活性,而错义突变或单个氨基酸的缺失可使VLCAD留有残余部分酶活性,症状较轻。研究还发现15个由于小缺失或小插入而导致的框移引起终止密码子早现的突变和9个剪切突变导致VLCAD残余酶活性的丧失,而导致严重的表现型。但Souri等[13]研究发现错义突变R573W与单个氨基酸缺失del258K却可导致VLCAD酶活性完全丧失。Coughlin等[18]对一生后38h急性死亡的VLCADD患儿研究发现此患儿存在错义突变V283A,而此突变在之前许多研究中发现存在残余酶活性,且在新生儿筛查阳性儿童和高危家庭筛查中发现一些无VLCADD症状个体也存有V283A突变,提示基因型对预测携带突变的患者是否存在致命并发症的风险存在局限性。Ficicioglu等[19]也认为基因突变分析并不能准确预测疾病的严重程度。基因突变后的极长链酰基辅酶A脱氢酶的残余酶活性不仅仅与基因突变的类型有关,还可能受温度等外界条件的影响。Fukao等[20]对A416T和R450H突变的体外研究发现, A416T突变在30℃的残余酶活性为正常对照的20%,而在37℃为正常的10%;突变R450H在30℃的残余酶活性为正常对照的5%,而在37℃时则完全丧失残余酶活性。可见温度升高可使残余酶活性降低,这可能是当患者处于感染发热时临床表现较重的一个重要原因。Tucci等[21]通过对VLCAD基因敲除小鼠模型研究发现,空腹可引起小鼠肝脏脂肪酸及中间代谢产物异常蓄积,导致过氧化物酶体和微粒体氧化通路酶表达上调,从而使线粒体内活性氧(ROS)大量生成及脂质过氧化物形成,引起细胞膜和线粒体的结构和功能障碍,最终导致肝脏脂肪变性,可见线粒体氧化应激和损伤可能为VLCADD发病机制之一。4 VLCADD的实验室检查及诊断 4.1 常规实验室检查可有低酮性低血糖,急性发作时可有代谢性酸中毒,肌酸激酶(CK)、肌酸激酶同工酶(CK-MB)及乳酸脱氢酶(LDH)水平升高,天冬氨酸氨基转移酶(AST)、丙氨酸氨基转移酶(ALT)水平升高。肌病型患者可有肌红蛋白尿,尿常规异常或伴有肾功能异常。肌活检可发现肌肉组织中有大量脂滴蓄积于Ⅰ型肌纤维。4.2 串联质谱检测血酰基肉碱谱可发现有多种长链酰基肉碱谱水平升高,其中以肉豆蔻烯酰基肉碱(C14:1)升高最为明显,因此将此项指标作为诊断极长链酰基肉碱辅酶A脱氢酶缺乏症最重要的代谢指标[22]。Liebig等[23]认为MS/MS进行新生儿疾病筛查中,若C14:1大于1umol/L可诊断为VLCADD患者。除此项指标外,可伴有C14:1/C10、C14、C14:2、C16、C18:1等多种长链酰基肉碱水平升高,C14:1/C10比值升高也同样有诊断意义[2]。由于线粒体内蓄积的C14-C18酰基辅酶A需与游离肉碱结合形成酰基肉碱而离开线粒体,VLACDD患者同时也存在组织中游离肉碱水平降低。进行尿气相质谱有机酸分析可发现二羧酸尿症,可有己二酸、辛二酸、癸二酸、十二烷二酸等水平升高,但轻症患者或甚至伴有横纹肌溶解患者可无二羧酸尿症[20]。4.3 酶学分析 可对患者的皮肤成纤维细胞、外周血淋巴细胞、心肌和骨骼肌细胞或组织进行极长链酰基辅酶A脱氢酶活性测定明确诊断。具体方法:提取细胞线粒体膜蛋白,加入特定底物棕榈酰CoA(16碳),反应后用高压液相色谱分析(HPLC)检测棕榈酰基肉碱(C16)、棕榈烯酰基肉碱( C16:1)及3-羟基棕榈烯酰基肉碱(C16:OH)浓度,反映极长链酰基辅酶A脱氢酶对棕榈酰CoA的氧化作用,测定反应速率,计算VLCAD酶活性[24]。4.4 脂肪酸β氧化流量分析 培养患者的皮肤成纤维细胞,分别加入用3H标记的[9,10(n)-3H]肉豆蔻酸酯(C14:0)和[9,10(n)-3H]棕榈酸酯(C16:0),通过测定产物3H2O的产生量,反映细胞中VLCAD对肉豆蔻酸酯和棕榈酰酯的氧化率[25]。虽然酶学分析和脂肪酸氧化流量分析对VLCADD有确诊意义,但检测复杂,尚未常规普及。4.5 基因诊断 基因突变分析也是确诊VLCADD的金标准,通过对ACADVL的20个外显子设计引物进行聚合酶链反应(PCR)和DNA测序寻找突变以明确诊断。对新生儿筛查发现C14:1升高但小于1umo/L诊断含糊的患儿可行此检测明确诊断[23]。5 VLCADD的治疗和预后 VLCADD总的治疗原则是避免空腹,高碳水化合物和低脂饮食尤其是限制长链脂肪酸的摄入,补充中链甘油三酯(Medium Chain Triglycerides, MCT),对症处理及预防和治疗并发症。5.1 避免空腹 新生儿患者一般间隔3h喂养一次;<6月婴儿间隔4h;6-12月婴儿夜间可间隔6-8h;1-7岁的儿童白天间隔4h,夜间可延长10h喂养;而成人一般间隔8小时(4-12h)。可在夜间或紧张活动时给予生玉米淀粉以加强对空腹的耐受,生玉米淀粉可持续释放葡萄糖,减少低血糖发生和脂肪的分解动员[26, 27]。 5.2 合理饮食和中链甘油三酯(MCT)的使用有症状的VLCADD患者脂肪摄入占总热卡的25-30%,尤其注意限制长链脂肪酸和补充MCT[27]。MCT由辛酸(8碳)及癸酸(10碳)等偶数碳的中链脂肪酸为主要成分构成的甘油三酯,这些脂肪酸的代谢不依赖于VLCAD的催化,中链脂肪酸可以直接穿过线粒体膜,经过线粒体脂肪酸β氧化过程生成乙酰CoA及生成酮体为机体功能。心肌病型患儿,MCT比例应占总脂肪摄入的90%,而长链脂肪酸占10%。5.3 左旋肉碱 对于肉碱补充治疗脂肪酸β氧化障碍疾病一直存有争议。在VLCADD中,由于极长链脂肪酸的β氧化通路受阻,而导致线粒体内过多的长链脂肪酸蓄积,这些脂肪酸则需与游离肉碱结合形成酰基肉碱转运出线粒体,这将造成组织以致血中游离肉碱的缺乏,故补充肉碱可以维持血中游离肉碱水平的稳定。一般给予50-100mg/kg/d。左旋肉碱配合饮食治疗可以明显缓解VLCADD患者的心功能异常。短期应用可以促进酮体生成和减少空腹低血糖的发生,但过多则促进长链酰基肉碱的生成和蓄积,对机体产生毒性作用。Primassin等[31]通过建立VLCADD的模型小鼠研究发现,补充肉碱并不能阻止肌肉中游离肉碱的降低,尤其在运动后,反而造成骨骼肌中大量酰基肉碱蓄积和毒性作用。 5.4 其他治疗 (a)对于反复低血糖发作的患者可以静脉注射葡萄糖以纠正低血糖症状。(b)有研究发现过氧化物酶体增殖激活受体(PPAR)α的激活剂苯扎贝特能提高VLCADD细胞的脂肪酸氧化能力,并提高VLCADD患者细胞中VLCAD mRNA及蛋白的表达量,通过上调基因表达来提高突变体蛋白的酶活性,同时苯扎贝特还能减少具有毒性作用的长链酰基肉碱的生成[32, 33]。(c)文献报道肌松剂丹曲洛林钠盐对伴有肌痛性痉挛、肌强直、横纹肌溶解的成人VLCADD患者具有良好的效果,主要机制是丹曲洛林钠盐能够结合骨骼肌肌浆网的主要Ca2+释放通道Ryandonine受体,限制Ca2+从肌质网/肌浆网中释放,并阻止细胞内Ca2+持续升高,及Ca2+升高引起的线粒体功能异常等;还可作用于神经肌肉接头使兴奋-收缩偶联中断[34],以达到治疗目的。综上所述,对极长链酰基辅酶A脱氢酶缺乏症的诊断和治疗尚需进一步的随访和研究,对于临床疑诊患者应尽早行血串联质谱和尿气相质谱等相关检查,对临床诊断的患者可行分子生物学或酶学及脂肪酸氧化流量分析以明确诊断,不同临床分型预后明显不同,明确基因诊断的基础上可为VLCADD患者家庭提供产前诊断和遗传咨询。对该病的预防需积极开展新生儿筛查,早期诊断,早期治疗可明显改善预后,提高患者生存率和生活质量。
邱文娟 主任医师 上海新华医院 儿童内分泌遗传科1.7万人已读 - 精选 小儿持续高热不退之原因之一:川崎病
川崎病是50多年前由一名日本儿科医生名字命名的一种疾病。是一种临床表现特殊,治疗方法特殊,需要长期随访,可导致小儿心脏病的特殊疾病。其特殊的临床表现主要包括:小儿持续高热,眼睛红,皮疹,口唇红干燥皲裂,草莓舌,手足硬,肿胀及颈部淋巴结肿大这些表现。该病的诊断主要依靠上述的临床表现,但需提醒大家的是并不是孩子得了川崎病上面的表现都会有,有的孩子仅表现为发热伴皮疹,眼红症状或其他一两个表现,这就是不典型川崎病。得了川崎病最可怕的是该病会导致小儿的心脏冠状动脉发炎,导致冠状动脉瘤的严重并发症。因此对于该病需及时诊断及治疗。一般来说在患儿发病的10天内用上治疗川崎病的药物即可预防患儿出现冠状动脉瘤,使其发生率由30%降低至5%左右。而治疗的最佳时间是发病的5-7天。因此对于你的宝宝如果出现持续发热不退,又出现了皮疹、或眼睛红、或脖子肿大、嘴唇红裂、草莓舌等表现,一定要及时就诊,由医生决定患儿是否患了川崎病,否则留下冠状动脉并发症就很严重了。
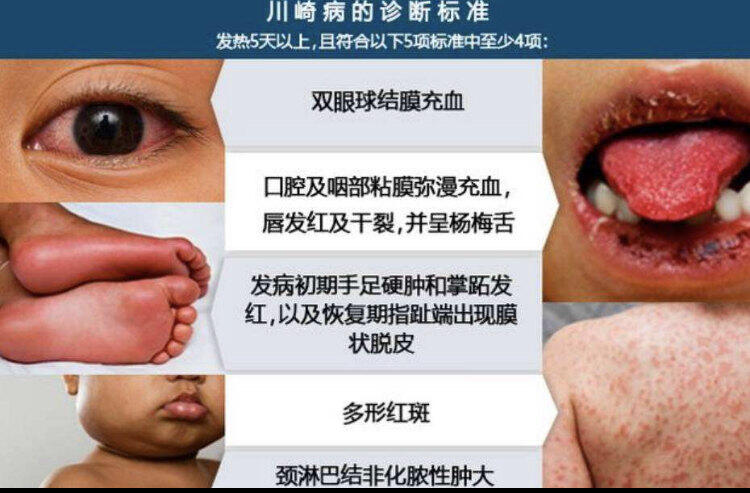 张清友 主任医师 北京大学第一医院 小儿心内科1700人已读
张清友 主任医师 北京大学第一医院 小儿心内科1700人已读 - 精选 小儿厌食,家长如何应对
什么是小儿厌食?小儿厌食症是什么?小儿厌食症指的是孩童长时间出现食欲减退或食欲不振,多发于1岁到6岁的孩童身上。一般来说,小儿厌食症的主要症状有很多,包括呕吐、食欲减退或食欲不振、便秘、腹痛腹泻和便血等症状。小儿厌食症多由不良的饮食习惯引起,微量元素的缺乏和多种慢性疾病等都会导致小儿厌食。‘ 小儿厌食的原因?1、多种急、慢性疾病常常伴有厌食。全身性疾病如结核病、胶原病、贫血及一些慢性感染等,胃肠道疾病如消化性溃疡、急慢性肝炎、慢性肠炎,肝功能不全、高血压、酸中毒、尿毒症、心功能不全以及消化道淤血,以及各种原因的腹泻及慢性便秘等都是常见的原因。 2、大多数的厌食症与不良的饮食习惯有关。零食过多、餐前饮用大量饮料、进食时注意力不集中(如边听故事、边看电视边吃饭)等等不良的习惯,可以扰乱或抑制胃酸及消化酶的分泌,从而使患儿食欲减退 3、家长长期强迫进食的恶果。这些家长他们常常过分担心小儿营养不足,体重增长不快,进食量过小等,强迫小儿进食。大大影响了小儿的情绪,产生了“进食等于受罪”的错觉,并逐渐形成了条件反射性拒食,最终发展成厌食。 4、长期服用药物可能会导致小儿厌食。如红霉素、氯霉素、磺胺类药物以及氨茶碱等。维生素A或维生素D服用过量导致中毒,也会引起小儿厌食。 小儿厌食,家长怎样预防1、要培养良好的饮食习惯,注意进食定时定量,一般儿童每日3餐,每餐间隔4-5小时,幼儿4餐间隔3-4小时;用餐时间最好不要超过半小时,不吃的话等用餐时间结束,所有食物收掉,二餐之间即使想吃也不能给食物,注重培养小孩自己的进食能力;这么做是孩子有空腹感,有利促进胃液的正常分泌。 2、控制零食:数量要控制,不可因贪吃零食而影响正餐摄入。优选零食品种。质量成分要讲究:上午宜给一点高热量食品,如巧克力、蛋糕、饼干等;午睡后喝点白开水,下午给一点水果 (在游戏的间歇期给予),晚餐后一般不再给零食,若有条件可在临睡前喝一杯牛奶。 3、安排食谱要力求多样化,让孩子有充分的选择余地,即可收到良好的效果。对偏食的孩子在改变烹调和家长的示范鼓励行为方面下点功夫,逐渐唤起孩子对食物的热情。 4、环境适宜:创造良好的就餐氛围,大人小孩一起吃,切忌吃饭时训斥或逗孩子玩;喂养得当宝宝不吃时不要追着喂,能吃多少算多少,避免伤食。 5、体育锻炼:在条件允许的情况下,增加孩子的户外活动量,以促进脾胃蠕动,促进食物消化。 6、用药要小心,不要滥用清热泻火类药物,如板蓝根冲剂、清热泻火口服液等,因为此类药物多性味苦寒伤胃;家长要切记,切不可因宝宝大便干结俗称“上火”,而过食寒凉类药物。不滥用抗生素;家庭用药可用益生菌,参苓白术散、婴儿健脾散、小儿化积口服液等中成药。 本文系曹飞跃医生授权好大夫在线(www.haodf.com)发布,未经授权请勿转载。
曹飞跃 副主任医师 娄星区人民医院 儿科1.2万人已读 - 精选 小儿夜遗尿的诊治
睡觉不尿床,孩子更快乐——小儿夜遗尿的诊治问题:请您介绍一下什么叫做夜间遗尿?答::夜间遗尿在医学上叫做遗尿症,俗称尿床。如果5岁以上,每个星期有两次以上的,在夜间睡眠中不自觉把尿排在床上,就可以诊断
徐心坦 主任医师 济宁医学院附属医院 儿科1.9万人已读 - 精选 孩子尿频是什么原因?
最近发现因为尿频来就诊的孩子多了起来,有女孩,也有男孩子;有的只有尿频,几分钟一次;有的有尿失禁(忍不住尿在身上);有的伴有夜间遗尿。泌尿系统很多疾病可以有尿频的症状,那么这些孩子到底是什么原因引起的呢? 总结了最近接诊的小病人,大约有如下几种情况: 1.泌尿道感染:有尿频表现,伴有尿失禁、遗尿的,还出现了外阴红肿痛、分泌物多、尿常规是大量白细胞尿的,属于最常见的泌尿道感染。泌尿道感染常常是通过尿培养检查来最终确定的,并且需要服用抗生素来治疗。 2.过敏性疾病:孩子尿频非常严重,伴有外阴红、骚痒,但没有分泌物,也没有尿常规检查异常。这样的孩子经常伴有长期咳嗽、流鼻涕、打鼾、皮肤红疹等等全身过敏症状。有的妈妈带孩子来不说明孩子还有这些问题,只管尿频,所以可能造成误诊。 3.神经性尿频:除外尿频,没有其它不适。很多妈妈十分焦急,却不知道是什么原因,总是想控制孩子排尿,结果适得其反。孩子的生活环境改变、与陌生人接触、强迫孩子做不愿做的事情,孩子与同学、老师之间的小磨擦,假期结束恢复上学等是可能的促发因素,尿频可能是孩子心理压力的反映。 4.寄生虫感染:有尿频、肛门骚痒、遗尿等表现。常见于幼儿园期小朋友,蛲虫感染时,可以在尿频、述屁股痒时检查一下肛门周围,看是否有虫体活动。如果是这个问题,应该以驱虫药治疗。 5.肾小球肾炎:除了尿频,可能还有更严重的表现,比如脸肿、尿少、茶色尿、蛋白尿等。应该尽快到医院肾脏专科就诊。 其实,诊断疾病需要很多的线索,希望爸爸妈妈们在百忙中多关心宝宝,这样可以尽早的发现孩子的病情,给予及时诊治和恰当的反映处理,是可以尽快的帮助孩子康复的。
沈彤 主任医师 厦门市妇幼保健院 儿科14.8万人已读 - 精选 孩子长期咳嗽要小心哪些病
在临床工作中,我们经常会遇见一些长期咳嗽(一个月以上)的孩子,家长们总以为孩子是感冒后嗓子发炎引起的咳嗽,于是断断续续的给孩子不断的吃不同的抗生素,甚至不停的给孩子输液,可是总感觉不能去“根”,不久,
刘海燕 副主任医师 西安交大二附院 小儿内科32.1万人已读 - 精选 对牛奶蛋白过敏的小孩转奶的相关问题
今天遇到一位家长向我讲述了她的就诊经历。小孩现在6月龄,4个月前因腹泻、大便带血丝就诊,在儿科门诊当成细菌性肠炎,使用抗生素治疗10天无效。后来在我这里就诊时,考虑为牛奶过敏(CMPA)所致。建议停抗生素,口服益生菌,并更换为水解配方奶粉喂养。大约1周后患儿腹泻好了,大便正常了,皮肤光滑了,体重增加也达标了。现在,这位妈妈不知道何时换回普通奶粉,又来咨询我。我想,牛奶蛋白过敏(CMPA)的患儿家属,可能都有这样的问题:我家宝宝何时能换回普通奶粉啊?甚至,某些家属认为自己宝宝过敏症状一消失,就可以尝试转成普通奶粉。今天我要提醒一下大家,过敏症状缓解不代表治愈!因为儿童的免疫功能是在不断成熟的过程当中,提前更换氨基酸配方奶粉,可以导致过敏症状长期反复,影响患儿生长发育,同时也增加了将来患过敏性鼻炎和支气管哮喘等疾病的风险。上图表示,0-18月婴幼儿如果不存在食物过敏、短暂食物过敏和长期食物过敏三种情况,以后(>2岁)过敏性鼻炎和支气管哮喘的发病率之间的比较。可以看出,长期食物过敏的患儿发展成为过敏性鼻炎和支气管哮喘的风险是短暂食物过敏患儿的3.4倍和5.5倍,是无食物过敏的患儿的4.7倍和10.6倍。所以,牛奶蛋白过敏(CMPA)的小孩,并不是随意可以转奶的。 问题一、怎样从深度水解配方奶转到普通配方奶呢? 如果已经吃深度水解配方满3个月的宝宝,过敏症状消失,无反复,而且满6月龄添加辅食顺利时,则可以尝试转普通配方奶。如果尝试失败,则需要继续喂养深度水解配方至少3个月。 问题二、如何从氨基酸奶粉转到深度水解配方奶粉呢? 过敏症状消失,无反复,且满6月龄辅食添加顺利时,可以逐渐转奶。所谓逐渐,可以参看下图: 记住哦,如果氨基酸奶转深度水解奶失败,得重来哦。继续喂养氨基酸奶至少3个月,再尝试转奶! 问题三、氨基酸奶粉有苦味,小孩不吃怎么办? 这是几乎所有家长都遇到过的问题。目前来说,还没有更好的方法让小孩喜欢上氨基酸奶粉的味道。这主要是一个习惯问题。就如吃惯母乳的婴儿(哪怕是用奶瓶喂养),要换成普通配方奶粉喂养,很多婴儿也有一个适应的过程。可以尝试在氨基酸奶粉中添加适量葡萄糖(非蔗糖!!!)以提高甜味,但不鼓励一直这样。一旦小孩适应氨基酸奶粉的口味后,尽早停止添加葡萄糖。 问题四、感觉小孩吃氨基酸奶粉体重增加慢,是不是氨基酸奶粉营养不够啊? 氨基酸奶粉提供的热卡与一般普通奶粉相差无几。因为奶粉中的蛋白质已经是氨基酸了,不需要肠道消化成氨基酸再吸收,所以小孩可能会有大便变少、排便间隔时间延长等情况。只要喂养量是足的,不会影响儿童生长发育,反而能够让牛奶蛋白过敏的小孩体重增加达标。*更多精彩文章,请关注江医生微信公众号:育儿芝士堡
江南 主治医师 医生集团-上海 儿科7130人已读 - 精选 支原体肺炎,怎么样才算治好了(有科普视频)?
区卫健委“孕妇课堂”上,儿童病情居家观察、预防及生长发育科普视频公开课https://wx.vzan.com/live/tvchat-1361998145?shareuid=132685620&vprid=0&sharetstamp=1611568780229得了支原体肺炎的孩子服药一段时间后,不咳嗽也不发烧了,是不是病就好了呢,能不能不吃药了呢?孩子到底有没有痊愈,让家长很不放心,带着这部分家长的问题,好大夫在线请教了复旦大学附属中山医院青浦分院儿科的徐灵敏大夫。孩子不再发烧咳嗽,就是治好了?徐灵敏大夫指出,小儿支原体肺炎的治疗与一般肺炎的治疗原则基本相同,采取综合治疗措施。包括一般治疗、对症治疗、抗生素的应用、肾上腺皮质激素,以及肺外并发症的治疗等5个方面。对于抗感染治疗,鉴于支原体细胞内寄生的生物学特性,疗程一般主张不少于2~3周,通常是3~6周,也有疗程更长的,停药过早易于复发。轻者分次口服治疗即可,重症可考虑静脉给药,临床医生一般的原则是,临床治愈后再口服抗生素巩固治疗2周。复诊发现体温正常,呼吸没有困难;听诊时肺里面没有太多湿罗音,并且没有喘息声,可以认为是疾病基本治好了。但是,患儿的呼吸系统中可能会有一定的支原体病原体遗留,这部分遗留会刺激呼吸道导致咳嗽;另外,呼吸道黏膜在修复过程中,腺体分泌会比正常情况下多一点,加上遗留支原体的刺激作用,孩子嗓子里还是会有痰排出。只要咳嗽、排痰不影响孩子的正常生活,体温、炎症指标正常,就可以停止治疗,等待症状逐渐康复就可以了。IgM指标不是评判疾病治愈的依据IgM抗体滴度是确诊支原体肺炎的重要指标,但不用于评判疾病是否治愈。徐灵敏大夫解释,抗体就像是人体免疫系统产生的针对某一类病原体的军队,如果病原体侵袭到机体里面,机体就会产生相应的抗体,用于对抗病原体,就像召集守城的士兵一样;病原体消除,士兵就会逐渐解散,数量慢慢减少。IgM抗体滴度的高低,跟疾病的严重程度有一定相关性,但不完全同步,临床上评估支原体肺炎是否治愈,并不依据这个指标。大多数情况下,IgM的抗体滴度于病原体完全清除两周以后才会正常。因此该指标过高时不必过于纠结,但可以作为观察评估支原体停止复制的依据。孩子多久可以回幼儿园?当孩子停止输液并且白细胞值、血沉血象正常后,一般还需要口服抗生素如阿奇霉素继续治疗两周。一般情况下输液结束后在家休息一周,之后就可以去幼儿园了。得过这种病是不是很容易再感染?大部分成年人呼吸道中都带有支原体,这些支原体通过日常接触,易于侵入孩子的呼吸道组织,如果抵抗力低下,支原体肺炎可能还会复发。疾病再发与环境中的病原体存有量,还有自身对病原体的敏感性,自身抵抗力都有很大关系。支原体一旦进入肺组织细胞内,是难以被清除掉的。应用抗生素治疗,比如红霉素或者阿奇霉素,只能把血液中的支原体清除掉;而对存在于组织细胞内的支原体效果很小,因此抵抗力低下的时候,这部分支原体可能会繁殖进入血液,导致疾病复发。但从另一个方面来说,一旦支原体在体内繁殖,诱发机体产生了IgM抗体,就会对支原体有一定的免疫作用,相当于体内已经存在针对这个病原体的军队了,对疾病的抵抗力反而会强一些。如果孩子的支原体肺炎刚刚治好,但近期又出现咳嗽发烧,是不是复发了?徐灵敏大夫指出,此时发现孩子发热,建议家长先在家里给孩子服用阿奇霉素3天,效果不明显后再就诊评估。因为除了针对支原体,阿奇霉素对其他的病原体也有抑制作用。支原体IgM抗体滴度升高是支原体复发的指标之一。【总结临床经验撰写科普文章是为了促进儿童健康,得不到利益收入的,出版科普图书《儿科常见病解惑》http://t.cn/RFTuddE也是如此,这是7折销售的连接,17.1元,购买后读书能得到更多育儿知识,也有助于培养儿童看书学习的习惯。】本文系徐灵敏医生授权好大夫在线(www.haodf.com)发布,未经授权请勿转载。
徐灵敏 主任医师 中山医院青浦分院 儿科25.5万人已读 - 精选 小儿心肌损伤不必过度惊慌
我们常常见到有的家长为孩子心肌损伤而焦急,其实完全没有必要。所谓心肌损伤是指心肌酶有不同程度的升高而心电图正常,临床上够不到诊断心肌炎的标准而做出的临时性诊断,并非一单独的疾病,往往伴随着其他疾病,如
王振先 主任医师 山东大学第二医院 儿内科27.8万人已读

管丽医师
常德市第一人民医院儿科
